I have taken the dataset from pentacene CSP (supplementary data from J. E. Campbell, J. Yang and G. M. DayPredicted energy–structure–function maps for the evaluation of small molecule organic semiconductors). Together 586 structures. Next I have computed minimum distance (2-norm) between pentacene molecules within every structure (0.5573). (The van der Walls radii are not good because some pentacene molecules in the structures overlap when the balls have this radius). The length of the covalent bond between two adjacent carbons is 1.3618 Angstroms. Based on this I have created a point cloud with 1108 points on a sphere around every atom of pentacene with the radius 0.5573/2.
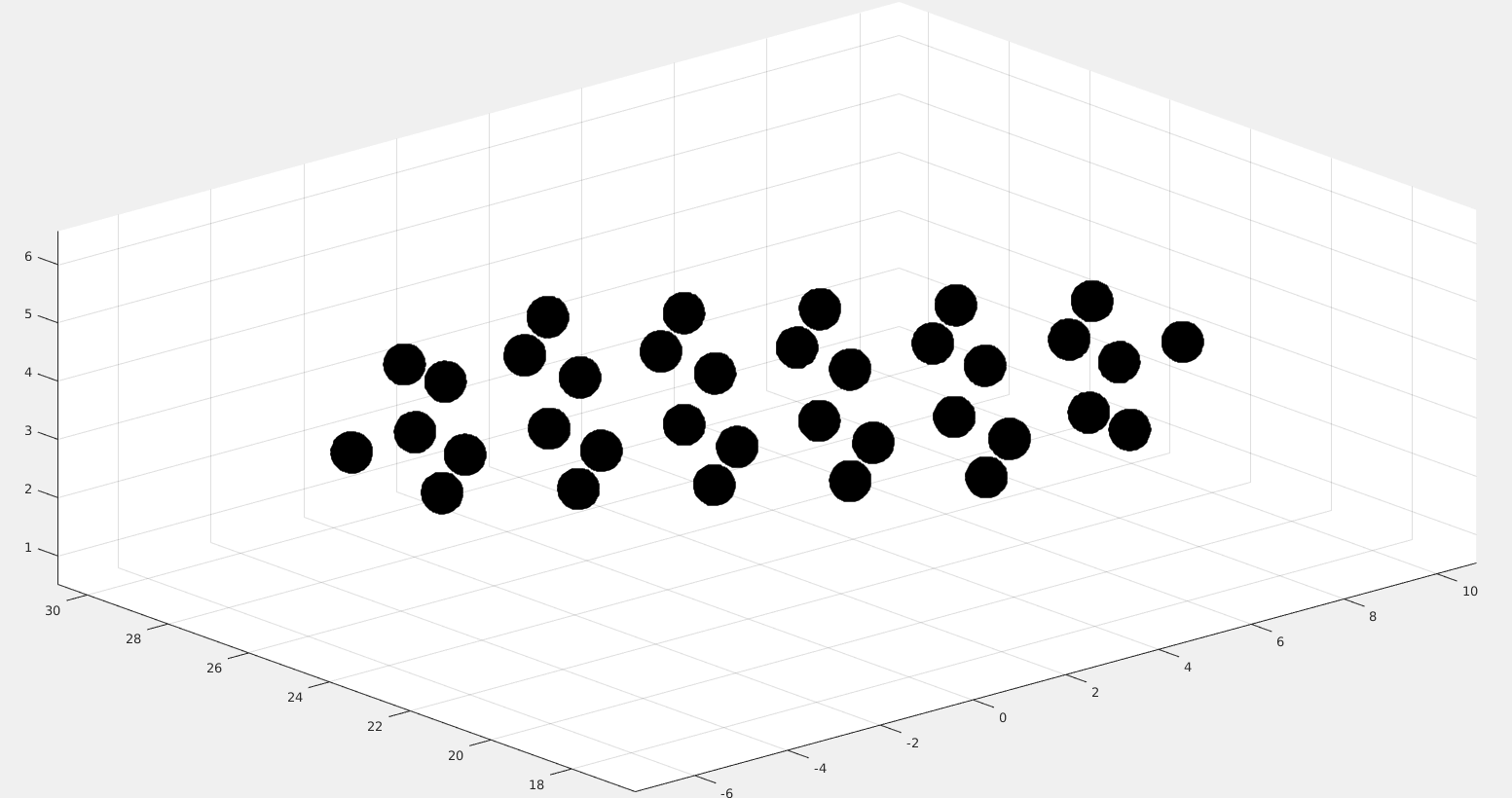
Below are the visualizations of convex hulls of the point cloud for 2 structures. On the left is the structure where the mentioned minimum is attained.
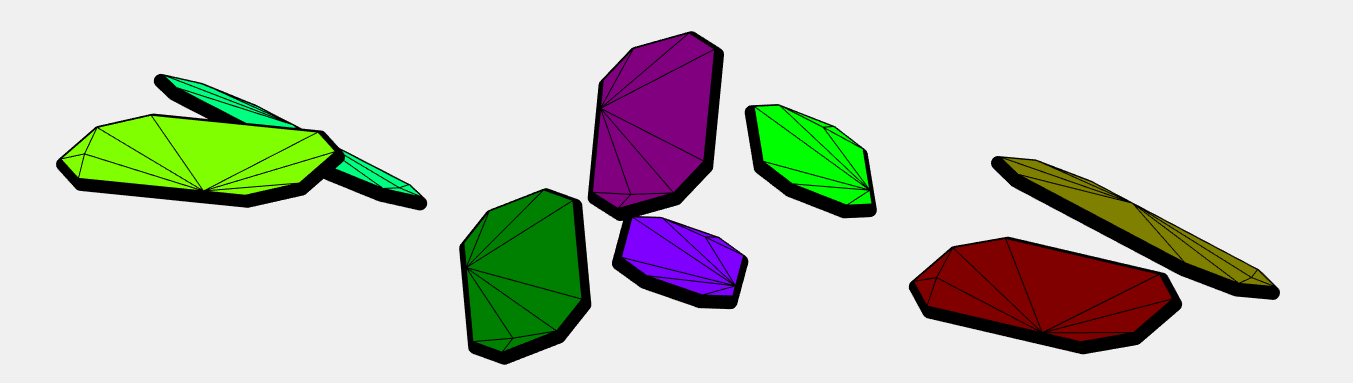
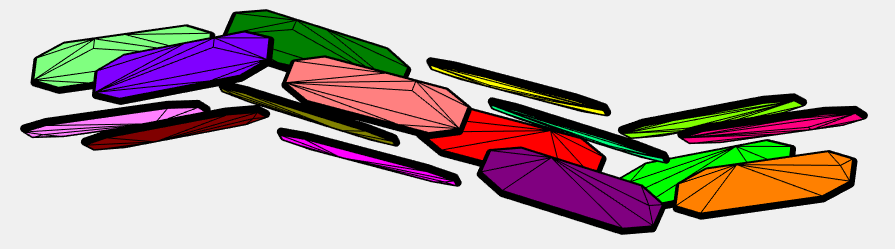
The O’Rourke’s minimum bounding box approach is not bad either, but we have to make the point could radius even smaller.
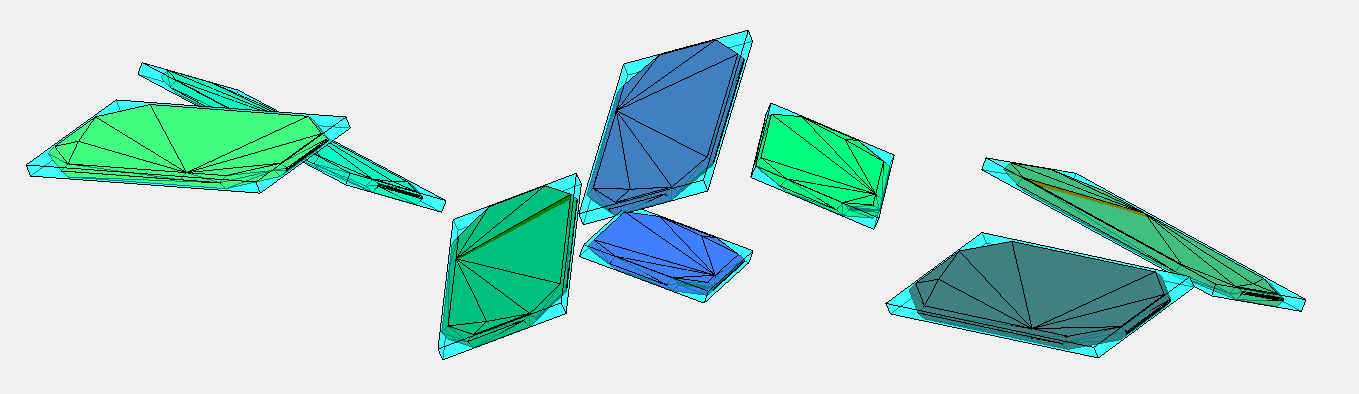
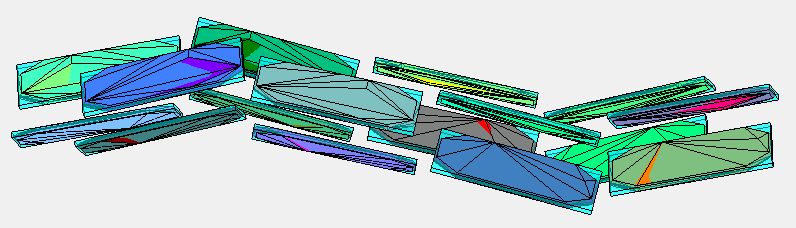
Conclusion: I think we should work with convex hulls.
Comment 1. : I discovered a stupid error I made with the minimal enclosing boxes modeling. Here it’s corrected but all the previous ones are most likely wrong.
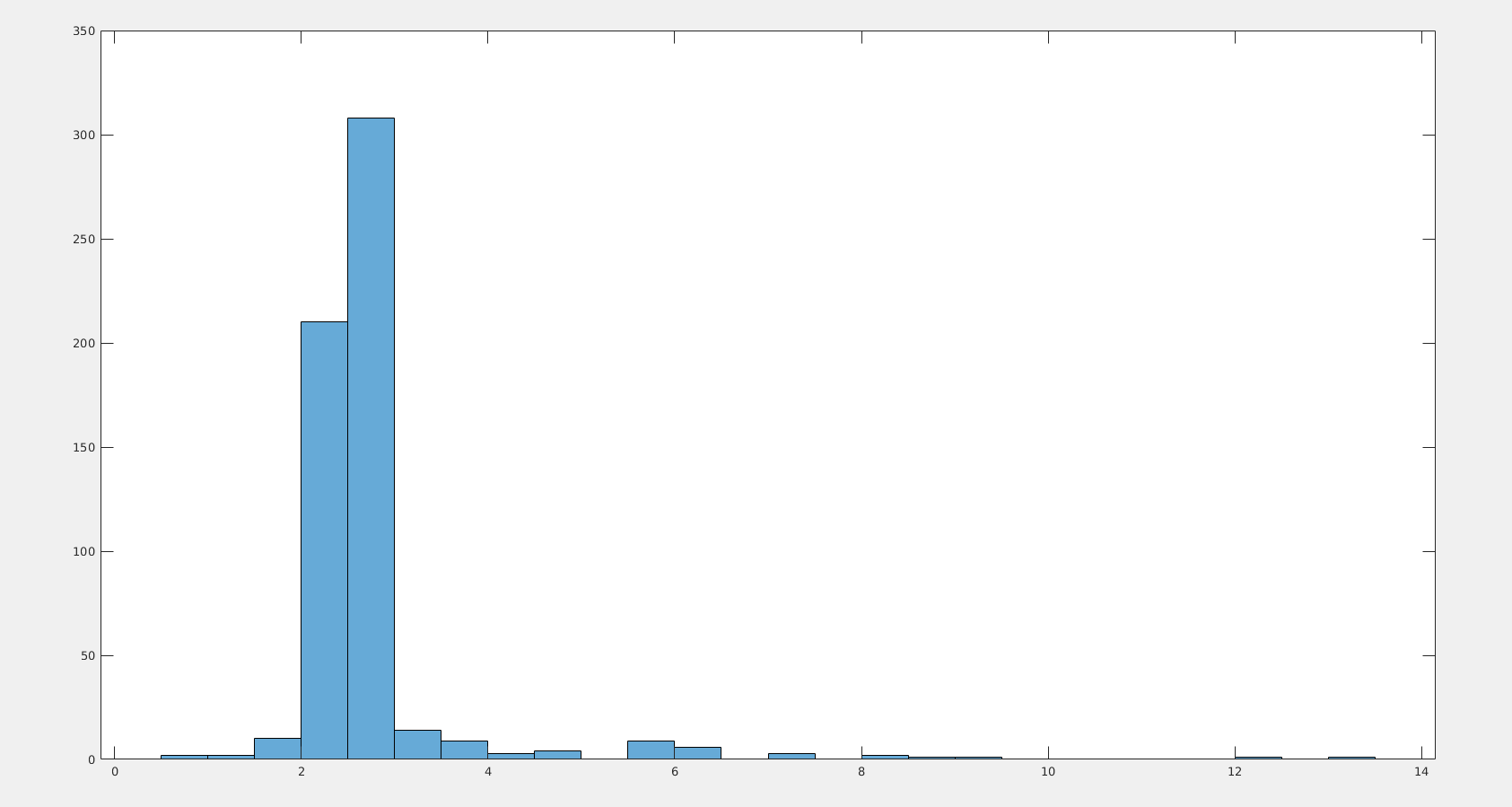
Comment 2. : the median of the minimum distances between pentacene molecules within the 586 structures is 2.6197. The histogram of the minima is below.